MLST¶
MLST (multi-locus sequence typing) was first developed in 1998 for N. meningitidis. The idea is to select a set of loci in the genome, and get the alleles in each genome for these loci. Typing is then done by seeing what isolates have which combination of alleles for each locus.
Schema¶
With MLST, the focus is on a subset of genes or loci that are known to be present in most, if not all strains of that species, but which are also known to have some sequence variation. The loci selected for use in MLST constitute the schema for that species. Commonly, for the original type of MLST, the number of genes chosen was 7. These are commonly housekeeping genes. There are several extensions of this that have come in later years, among them core genome MLST (cgMLST) and ribosomal MLST (rMLST). The main difference between the methods is in the criteria for how loci are included in the schema.
Allele and nomenclature¶
For each gene or locus in the schema, it is possible to have different sequence variants, or alleles. The different variants that have been observed can be collected in a database. Each observed allele for a gene can in this database be given a label, commonly a number. It is important to realize that these labels or numbers are attributed sequentially when detected, but do NOT provide any indication of relatedness or similarity between the alleles.
For the original “7-gene” type of MLST, several international nomenclature databases with alleles have been established. With access to such nomenclature databases, it is possible to ensure that all alleles have a unique number. This means that if using the same schema and the same database, two institutions can exchange information and know that they are referring to the same alleles.
Profile and sequence type¶
Each isolate can be typed by finding the sequence for each of the loci the schema contains. Once the sequence is found, it is compared to known alleles for that locus. This is usually done by comparing to alleles downloaded or otherwise accessed from the nomenclature server. If it is identical to an allele from the nomenclature database, the allele number for that locus is assigned to that isolate. If not, the allele is commonly uploaded to the server, which then assigns it a new number. This process happens automatically in some tools, in other cases it is done manually. The set of identifiers for an isolate is called its profile. The profile is commonly collapsed into a sequence type number, where each ST represents a unique combination of alleles for each locus.
Provided that the allele numbers and sequence type numbers are assigned by the same central nomenclature server, sequence types can be compared between institutions. Some institutions, like EFSA, can under certain circumstances allow organizations to submit sequence types, thus avoiding the need for uploading the full sequence.
MLST resources¶
Typing/Nomenclature databases¶
There are three main MLST nomenclature/database servers.
- https://pubmlst.org/
- https://bigsdb.pasteur.fr/
- https://enterobase.warwick.ac.uk/
It is important to realize that these work independently from each other and may contain different schemas, and even if they have the same schema there is no guarantee that the allele numbering is the same. Thus a sequence type only makes sense provided it is known which system has been used.
MLST finding methods¶
There are two main strategies for finding MLST genes.
- Assembly strategy: in this case the genomes are first assembled. Next, the allele set included in the typing database is used to search the genome, commonly using BLAST. The results are subsequently post-processed to get the allele and then compared to a profile database to assign ST numbers.
- Mapping strategy: in this case, the reads for each isolate are mapped against the allele typing database. Next, those reads are collected and assembled into genes. These are then compared to the alleles in the database, and allele and ST numbers are assigned.
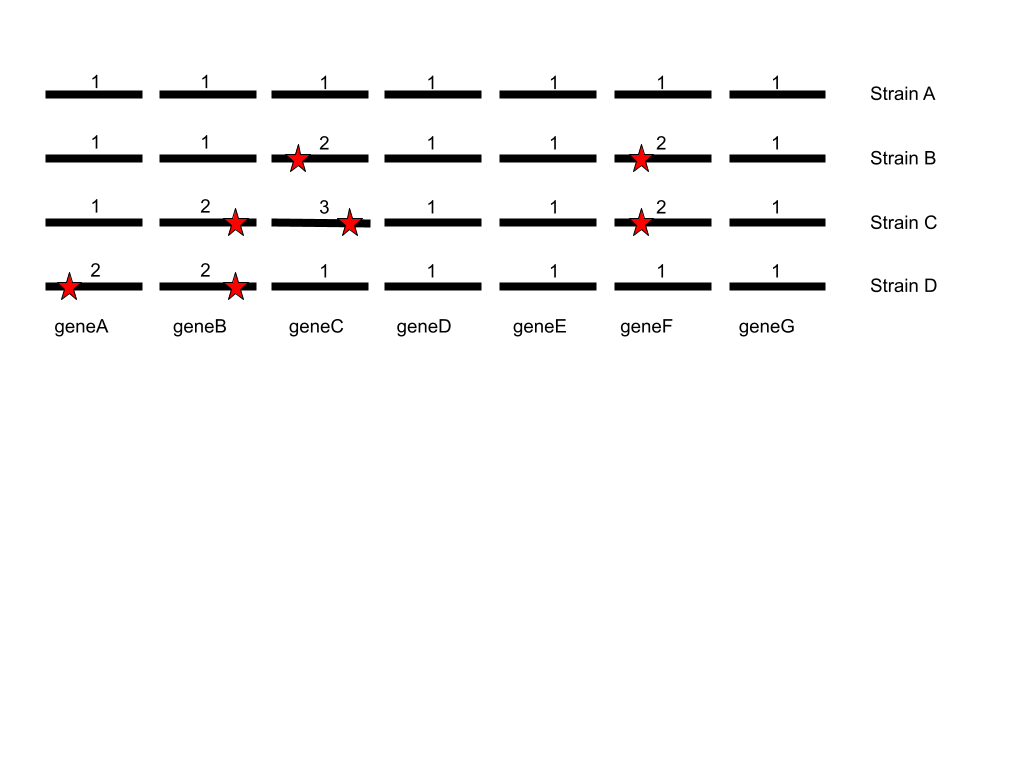 This figure shows how MLST works. In this case there are seven loci or genes
being used, genes A to G. Strain A is the first strain that is classified, thus
each version or allele of these genes are given the number 1. Subsequent new
alleles found in other strains are then given new numbers as they are found.
Note, the numbers are specific to each locus or gene, thus two different alleles
within the same strain can have the same number.
This figure shows how MLST works. In this case there are seven loci or genes
being used, genes A to G. Strain A is the first strain that is classified, thus
each version or allele of these genes are given the number 1. Subsequent new
alleles found in other strains are then given new numbers as they are found.
Note, the numbers are specific to each locus or gene, thus two different alleles
within the same strain can have the same number.
MLST software¶
There are many tools for doing MLST from both reads and assemblies available. It is important to realize that even though they might use the same schema, they might not give the same results. A review of various MLST tools and their results can be found in this paper. Due to this review no extensive examination of tools is given here. Some communities lean towards using certain tools, if so, these are mentioned in the Species specific sections.