Sequence analysis tool chains¶
In recent years next generation sequencing has matured and with that a solidification on the required methods for WGS projects has occurred. Nonetheless, the current practice of microbial WGS is mostly confined to the academic world, and has not yet resulted in large scale introduction of WGS in clinical settings, with the exception of a few countries. However, while it is clear that as the technological developments in bioinformatics proceed at a rapid pace, a consensus has formed on which steps are required to produce WGS datasets that can be used for surveillance and outbreak detection and investigation. These steps involve quality control of the raw sequence data and a subsequent usage of clean data to produce an annotated genome or reference map. Such “products” can then be used to obtain typing data, which is needed for the final analysis under surveillance or outbreak scenarios. These steps are illustrated in the figure below.
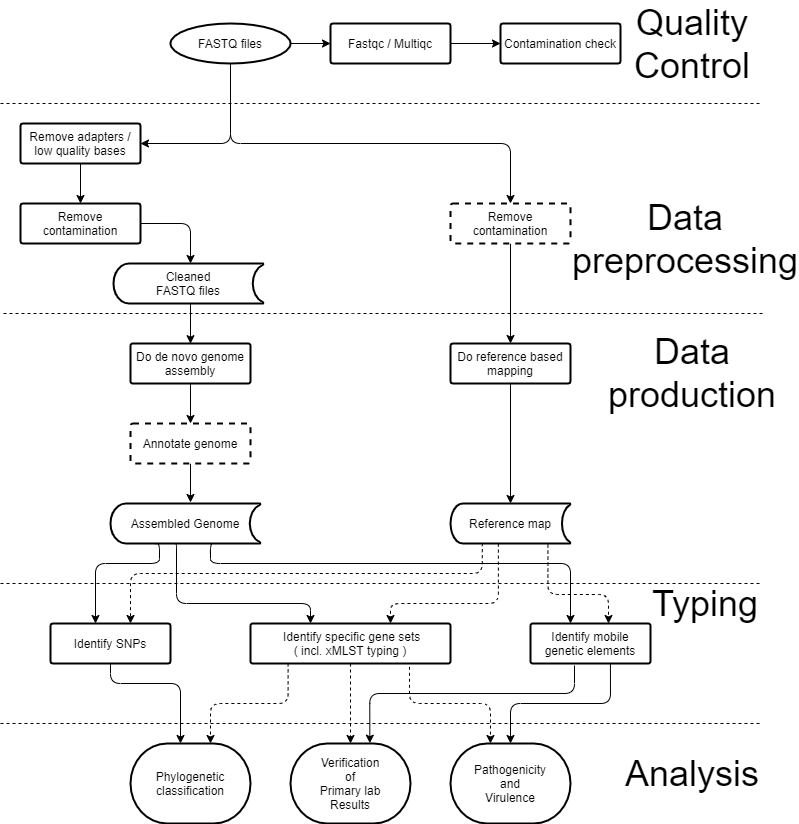 Flowchart showing a standardized WGS bioinformatic analysis workflow. The standardized workflow can be divided into four sections: Quality control, Data preprocessing, Data production, and Data analysis. Boxes with dotted lines are optional in the workflows presented. Solid and dotted lines are used for clarity and both indicate possible analysis pathways.
Flowchart showing a standardized WGS bioinformatic analysis workflow. The standardized workflow can be divided into four sections: Quality control, Data preprocessing, Data production, and Data analysis. Boxes with dotted lines are optional in the workflows presented. Solid and dotted lines are used for clarity and both indicate possible analysis pathways.
The first part of the process is to ensure that the quality of the reads is sufficient to proceed. Then, reads are ‘trimmed’, i.e. low quality parts and adapters are removed, and they are checked for any contamination. Once the sequence data is clean, it can then be used in two different downstream processes.
- Genome assembly, which is the computational process of combining all the shotgun DNA sequences in order to recreate the original genome sequence of an organism.
- Reference mapping, which is the alignment of the shotgun sequence fragments to a chosen reference genome, in order to identify differences between the reference and the investigated isolate.
Based on either a mapping or a genome assembly, the isolates can be further characterized through various typing and clustering tools. Different tools need different inputs - in most cases they either take an assembly or reads, but not both. It should be noted that the read mapping pathway is usually a lot less computationally expensive than the assembly pathway.
As is evident from the processes described here, the tool chain needed for analysis can get quite complicated. For these purposes it can often be useful to either use a workflow language, like Nextflow and Snakemake, or to use an analysis platform, like those described under Data management and analysis platforms. Without such systems it can get quite complicated to keep account of what analysis has been done, and the version of tools used.